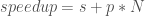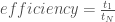In Parallel Programming Part 1, I introduced a number of basic OpenMP directives and constructs, as well as provided a quick guide to high-performance computing (HPC) terminology. In Part 2 (aka this post), we will apply some of the constructs previously learned to execute a few OpenMP examples. We will also view an example of incorrect use of OpenMP constructs and its implications.
TheCube Cluster Setup
Before we begin, there are a couple of steps to ensure that we will be able to run C++ code on the Cube. First, check the default version of the C++ compiler. This compiler essentially transforms your code into an executable file that can be easily understood and implemented by the Cube. You can do this by entering
g++ --version
into your command line. You should see that the Cube has G++ version 8.3.0 installed, as shown below.
Now, create two files:
openmp-demo-print.cpp this is the file where we will perform a print demo to show how tasks are distributed between threads
openmp-demo-access.cpp this file will demonstrate the importance of synchronization and the implications if proper synchronization is not implemented
Example 1: Hello from Threads
Open the openmp-demo-print.cpp file, and copy and paste the following code into it:
#include <iostream>
#include <omp.h>
#include <stdio.h>
int main() {
int num_threads = omp_get_max_threads(); // get max number of threads available on the Cube
omp_set_num_threads(num_threads);
printf("The CUBE has %d threads.\n", num_threads);
#pragma omp parallel
{
int thread_id = omp_get_thread_num();
printf("Hello from thread num %d\n", thread_id);
}
printf("Thread numbers written to file successfully. \n");
return 0;
}
Here is what the code is doing:
Lines 1 to 3 are the libraries that need to be included in this script to enable the use of OpenMP constructs and printing to the command line.
In main() on Lines 7 and 8, we first get the maximum number of threads that the Cube has available on one node and assign it to the num_threads variable. We then set the number of threads we want to use.
Next, we declare a parallel section using #pragma omp parallel beginning on Line 13. In this section, we are assigning the printf() function to each of the num_threads threads that we have set on Line 8. Here, each thread is now executing the printf() function independently of each other. Note that the threads are under no obligation to you to execute their tasks in ascending order.
Finally, we end the call to main() by returning 0
Be sure to first save this file. Once you have done this, enter the following into your command line:
This tells the system to compile openmp-demo-print.cpp and create an executable object using the same file name. Next, in the command line, enter:
./openmp-demo-print
You should see the following (or some version of it) appear in your command line:
Notice that the threads are all being printed out of order. This demonstrates that each thread takes a different amount of time to complete its task. If you re-run the following script, you will find that the order of threads that are printed out would have changed, showing that there is a degree of uncertainty associated with the amount of time they each require. This has implications for the order in which threads read, write, and access data, which will be demonstrated in the next example.
Example 2: Who’s (seg)Fault Is It?
In this example, we will first execute a script that stores each thread number ID into an array. Each thread then accesses its ID and prints it out into a text file called access-demo.txt. See the implementation below:
#include <iostream>
#include <omp.h>
#include <vector>
using namespace std;
void parallel_access_demo() {
vector<int> data;
int num_threads = omp_get_max_threads();
// Parallel region for appending elements to the vector
#pragma omp parallel for
for (int i = 0; i < num_threads; ++i) {
// Append the thread ID to the vector
data.push_back(omp_get_thread_num()); // Potential race condition here
}
}
int main() {
// Execute parallel access demo
parallel_access_demo();
printf("Code ran successfully. \n");
return 0;
}
As per the usual, here’s what’s going down:
After including the necessary libraries, we use include namespace std on Line 5 so that we can more easily print text without having to used std:: every time we attempt to write a statement to the output file
Starting on Line 7, we write a simple parallel_access_demo() function that does the following:
Initialize a vector of integers very creatively called data
Get the maximum number of threads available per node on the Cube
Declare a parallel for region where we iterate through all threads and push_back the thread ID number into the data vector
Finally, we execute this function in main() beginning Line 19.
If you execute this using a similar process shown in Example 1, you should see the following output in your command line:
Congratulations – you just encountered your first segmentation fault! Not to worry – this is by design. Take a few minutes to review the code above to identify why this might be the case.
Notice Line 15. Where, each thread is attempting to insert a new integer into the data vector. Since push_back() is being called in a parallel region, this means that multiple threads are attempting to modify data simultaneously. This will result in a race condition, where the size of data is changing unpredictably. There is a pretty simple fix to this, where we add a #pragma omp criticalbefore Line 15.
As mentioned in Part 1, #pragma omp critical ensures that each thread executes its task one at a time, and not all at once. It essentially serializes a portion of the parallel for loop and prevents race conditions by ensuring that the size of data changes in a predictable and controlled manner. An alternative would be to declare #pragma omp single in place of #pragma omp parallel foron Line 12. This essentially delegates the following for-loop to a single thread, forcing the loop to be completed in serial. Both of these options will result in the blissful outcome of
Summary
In this post, I introduced two examples to demonstrate the use of OpenMP constructs. In Example 1, I demonstrated a parallel printing task to show that threads do not all complete their tasks at the same time. Example 2 showed how the lack of synchronization can lead to segmentation faults due to race conditions, and two methods to remedy this. In Part 3, I will introduce GPU programming using the Python PyTorch library and Google Colaboratory cloud infrastructure. Until then, happy coding!
In my recent Applied High Performance Computing (HPC) lectures, I have been learning to better utilize OpenMP constructs and functions . If these terms mean little to nothing to you, welcome to the boat – it’ll be a collaborative learning experience. For full disclosure, this blog post assumes some basic knowledge of parallel programming and its related terminology. But just in case, here is a quick glossary:
Node: One computing unit within a supercomputing cluster. Can be thought of as on CPU unit on a traditional desktop/laptop.
Core: The physical processing unit within the CPU that enables computations to be executed. For example, an 11th-Gen Intel i5 CPU has four cores.
Processors/threads: Simple, lightweight “tasks” that can be executed within a core. Using the same example as above, this Intel i5 CPU has eight threads, indicating that each of its cores can run two threads simultaneously.
Shared memoryarchitecture: This means that all cores share the same memory within a CPU, and are controlled by the same operating system. This architecture is commonly found in your run-of-the-mill laptops and desktops.
Distributed memory architecture: Multiple cores run their processes independently of each other. They communicate and exchange information via a network. This architecture is more commonly found in supercomputer clusters.
Serial execution: Tasks and computations are completed one at a time. Typically the safest and most error-resistant form of executing tasks, but is the least efficient.
Parallel execution: Tasks and computations are completed simultaneously. Faster and more efficient than serial execution, but runs the risk of errors where tasks are completed out of order.
Synchronization: The coordination of multiple tasks or computations to ensure that all processes aligns with each other and abide by a certain order of execution. Synchronization takes up more time than computation. AN overly-cautious code execution with too many synchronization points can result in unnecessary slowdown. However, avoiding it altogether can result in erroneous output due to out-of-order computations.
Race condition: A race condition occurs when two threads attempt to access the same location on the shared memory at the same time. This is a common problem when attempting parallelized tasks on shared memory architectures.
Load balance: The distribution of work (tasks, computations, etc) across multiple threads. OpenMP implicitly assumes equal load balance across all its threads within a parallel region – an assumption that may not always hold true.
This is intended to be a two-part post where I will first elaborate on the OpenMP API and how to use basic OpenMP clauses to facilitate parallelization and code speedup. In the second post, I will provide a simple example where I will use OpenMP to delineate my code into serial and parallel regions, force serialization, and synchronize memory accesses and write to prevent errors resulting from writing to the same memory location simultaneously.
What is OpenMP?
OpenMP stands for “Open Specification for Multi-Processing”. It is an application programming interface (API) that enables C/C++ or Fortran code to “talk” (or interface) with multiple threads within and across multiple computing nodes on shared memory architectures, therefore allowing parallelism. It should not be confused with MPI (Message Passing Interface), which is used to develop parallel programs on distributed memory systems (for a detailed comparison, please refer to this set of Princeton University slides here). This means that unlike a standalone coding language like C/C++. Python, R, etc., it depends only on a fixed set of simple implementations using lightweight syntax (#pragma omp [clause 1] [clause 2]) . To begin using OpenMP, you should first understand what it can and cannot do.
Things you can do with OpenMP
Explicit delineation of serial and parallel regions
One of the first tasks that OpenMP excels at is the explicit delineation of your code into serial and parallel regions. For example:
In the code block above, we are delineating an entire parallel region where the task performed by the do_a_parallel_task() function is performed independently by each thread on a single node. The number of tasks that can be completed at any one time depends on the number of threads that are available on a single node.
Hide stack management
In addition, using OpenMP allows you toautomate memory allocation, or “hide stack management”. Most machines are able to read, write, and (de)allocate data to optimize performance, but often some help from the user is required to avoid errors caused by multiple memory access or writes . OpenMP allows the user to automate stack management without explicitly specifying memory reads or writes in certain cache locations via straightforward clauses such as collapse() (we will delve into the details of such clauses in later sections of this blog post).
Allow synchronization
OpenMP also provides synchronization constructs to enforce serial operations or wait times within parallel regionsto avoid conflicting or simultaneous read or write operations where doing so might result in erroneous memory access or segmentation faults. For example, a sequential task (such as a memory write to update the value in a specific location) that is not carefully performed in parallel could result in output errors or unexpected memory accessing behavior. Synchronization ensures that multiple threads have to wait until the last operation on the last thread is completed, or only one thread is allowed to perform an operation at a time. It also protects access to shared data to prevent multiple reads (false sharing). All this can be achieved via the critical, barrier, or single OpenMP clauses.
Creating a specific number of threads
Unless explicitly stated, the system will automatically select the number of threads (or processors) to use for a specific computing task. Note that while you can request for a certain number of threads in your environment using export OMP_NUM_THREADS, the number of threads assigned to a specific task is set by the system unless otherwise specified. OpenMP allows you to control the number of threads you want to use in a specific task in one of two ways: (a) using omp_set_num_threads() and using (b) #pragma omp parallel [insert num threadshere]. The former allows you to enforce an upper limit of the number of threads to be freely assigned to tasks throughout the code, while the latter allows you to specify a set number of threads to perform an operation(s) within a parallel region.
In the next section, we will list some commonly-used and helpful OpenMP constructs and provide examples of their use cases.
OpenMP constructs and their use cases
OMP_NUM_THREADS This is an environment variable that can be set either in the section of your code where you defined your global variables, or in the SLURM submission script you are using to execute your parallel code.
omp_get_num_threads() This function returns the number of threads currently executing the parallel region from which it is called. Use this to gauge how many active threads are being used within a parallel region
omp_set_num_threads() This function specifies the number of threads to use for any following parallel regions. It overrides the OMP_NUM_THREADS environment variable. Be careful to not exceed the maximum number of threads available across all your available cores. You will encounter an error otherwise.
omp_get_max_threads() To avoid potential errors from allocating too many threads, you can use omp_set_num_threads() with omp_get_max_threads(). This returns the maximum number of threads that can be used to form a new “team” of threads in a parallel region.
#pragma omp parallel This directive instructs your compiler to create a parallel region within which it instantiates a set (team) of threads to complete the computations within it. Without any further specifications, the parallelization of the code within this block will be automatically determined by the compiler.
#pragma omp for Use this directive to instruct the compiler to parallelize for-loop iterations within the team of threads that it has instantiated. This directive is often used in combination with #pragma omp parallel to form the #pragma omp parallel for construct which both creates a parallel region and distributes tasks across all threads.
To demonstrate:
#include <stdio.h>
#include <omp.h>
void some_function() {
#pragma omp parallel {
#pragma omp for
for (int i = 0; i < some_num; i++) {
perform_task_here();
}
}
}
is equivalent to
#include <stdio.h>
#include <omp.h>
void some_function() {
#pragma omp parallel for {
for (int i = 0; i < some_num; i++) {
perform_task_here();
}
}
#pragma omp barrier This directive marks the start of a synchronization point. A synchronization point can be thought of as a “gate” at which all threads must wait until all remaining threads have completed their individual computations. The threads in the parallel region beyond omp barrier will not be executed until all threads before is have completed all explicit tasks.
#pragma omp single This is the first of three synchronization directives we will explore in this blog post. This directive identifies a section of code, typically contained within “{ }”, that can only be run with one thread. This directive enforces serial execution, which can improve accuracy but decrease speed. The omp single directive imposes an implicit barrier where all threads after #pragma omp single will not be executed until the single thread running its tasks is done. This barrier can be removed by forming the #pragma omp single nowait construct.
#pragma omp master This directive is similar to that of the #pragma omp single directive, but chooses only the master thread to run the task (the thread responsible creating, managing and discarding all other threads). Unlike #pragma omp single, it does not have an implicit barrier, resulting in faster implementations. Both #pragma omp single and #pragma omp master will result in running only a section of code once, and it useful for controlling sections in code that require printing statements or indicating signals.
#pragma omp critical A section of code immediate following this directive can only be executed by one thread at a time. It should not be confused with #pragma omp single. The former requires that the code be executed by one thread at a time, and will be executed as many times as has been set byomp_set_num_threads(). The latter will ensure that its corresponding section of code be executed only once. Use this directive to prevent race conditions and enforce serial execution where one-at-a-time computations are required.
Things you cannot (and shouldn’t) do with OpenMP
As all good things come to an end, all HPC APIs must also have limitations. In this case, here is a quick rundown in the case of OpenMP:
OpenMP is limited to function on shared memory machines and should not be run on distributed memory architectures.
It also implicitly assumes equal load balance. Incorrectly making this assumption can lead to synchronization delays due to some threads taking significantly longer to complete their tasks compared to others immediately prior to a synchronization directive.
OpenMP may have lower parallel efficiency due to it functioning on shared memory architectures, eventually being limited by Amdahl’s law.
It relies heavily on parallelizable for-loops instead of atomic operations.
Summary
In this post, we introduced OpenMP and a few of its key constructs. We also walked through brief examples of their use cases, as well as limitations of the use of OpenMP. In our next blog post, we will provide a tutorial on how to write, compile, and run a parallel for-loop on Cornell’s Cube Cluster. Until then, feel free to take a tour through the Cornell Virtual Workshop course on OpenMP to get a head start!
If you’ve ever run into software dependency issues when trying to download and run a computer model on a new machine, you’re in good company. Dependency issues are a common headache for modelers, whether you are trying to run someone else’s code or share your own code with collaborators. Problems can arise due to differences in operating system (e.g., Windows vs OSX vs Ubuntu), compilers (e.g., Gnu vs Intel), software versions (e.g., Python 3.3 vs 3.10), or other factors. These challenges are exacerbated on high performance computing (HPC) clusters, which often have highly specialized architectures and software environments that are beyond our control. For example, I regularly develop code on my laptop, test parallel jobs on TheCube cluster at Cornell, and finally run much larger production jobs on the Stampede2 cluster at TACC. Ensuring compatibility across these three environments can be a challenge, especially for models involving complex software interdependencies and/or MPI parallel communication. Thankfully, containers offer an elegant solution to many of these challenges.
Containers such as Docker allow you to package an application in a self-contained, ready-to-run package. The container includes its own operating system and all libraries and dependencies needed to run your code. The application then runs inside this custom container where it is isolated from the individual machine that you are running it on, making it much simpler to build portable applications. This has several benefits for researchers by allowing you to spend more time on scientific research and model building and less time on installing finicky software dependencies. Additionally, it can encourage “open science” and collaboration by streamlining the process of sharing and borrowing code.
However, HPC clusters present some unique challenges to the “build once, run anywhere” mantra of containerization. First, Docker containers generally require root permissions that are disallowed on shared supercomputers. This challenge can be overcome by using a second type of container called Singularity. Second, when running parallel applications with MPI, or running applications on GPUs, the container must be built in a way that is consistent with the hardware and software setup of the cluster. This challenge is harder to address, and does reduce the portability of the container somewhat, but as we shall see it can still be overcome with careful container design.
This blogpost will demonstrate how to build containers to run parallelized code on HPC clusters. We will build two different containers: one which can be run on two different TACC clusters, Stampede2 and Frontera, and one which can be run on two different Cornell Center for Advanced Computing (CAC) clusters, TheCube and Hopper. Both of these can also be run on a personal laptop. If you want to follow along with this post and/or containerize your own code, the first thing you will need to do is to download and install Docker Desktop and set up an account on DockerHub. You can find instructions for doing that, as well as a lot more great information on containerization and HPC, in this documentation from the Texas Advanced Computing Center (TACC). You can also find more information on Docker in earlier Water Programming Blog posts by Lillian Lau, Rohini Gupta, and Charles Rouge.
Containerizing a Cython reservoir model with Docker
All code related to this blogpost can be found in the “WPB_containerization” directory of my “cython_tutorial” GitHub repository, here. If you want to follow along, you can clone this repository and navigate in your terminal to the WPB_containerization directory. This directory contains a simple reservoir simulation model which is built using Cython, a software used to convert Python-like code into typed, compiled C code, which can significantly improve execution time. Cython is beyond the scope of this blog post (though I may cover it in a future post, so stay tuned!), but the interested reader can find an overview in the base directory of this GitHub repository, and references therein. For this post, suffice it to say that Cython introduces non-trivial software dependencies and build complexities beyond a more standard Python application, which makes it a good candidate for containerization. Additionally, this directory contains scripts for running parallel simulations across multiple nodes on an HPC cluster using MPI, which introduces additional containerization challenges.
The first container we will build is for the TACC clusters Stampede2 and Frontera. The main step involved in building a Docker container is writing the Dockerfile (see the file “Dockerfile_tacc”), which outlines the steps needed to build the container:
FROM tacc/tacc-ubuntu18-impi19.0.7-common
RUN apt-get update && apt-get upgrade -y && apt-get install -y libjpeg-dev python3-pip
RUN pip3 install numpy pandas matplotlib cython mpi4py
ADD . /code WORKDIR /code
RUN python3 setup.py build_ext --inplace
RUN chmod +rx /code/run_reservoir_sim.py
ENV PATH "/code:$PATH" RUN useradd -u 8877 <user>
The first line tells Docker which “base image” to start from. There are a wide variety of base images to choose from, ranging from standard operating systems (e.g., Ubuntu 18.04) to smaller specialized images for software languages like Python. Due to the complexity of TACC’s hardware and software infrastructure, they provide special base images that are set up to efficiently interface with the clusters’ particular processors, compilers, communication networks, etc. The base image imported here is designed to operate on both Stampede2 and Frontera. In general, it should also work on your local computer as well, which helps streamline application development.
The second line updates the Ubuntu operating system and installs Python3 and another necessary library. Third, we install Python libraries needed for our application, including Cython and mpi4py. We then add the contents of the current working directory into the container, in a new directory called /code, and make this the working directory inside the container. Next, we run setup.py, which cythonizes and compiles our application. We then make the file “run_reservoir_sim.py” executable, so that we can run it directly from the container, and add the /code directory to the container’s path. Lastly, we change the user from root to <user> (fill in your user profile name on your computer) to avoid permissions issues on some machines.
where <username> should be replaced with your DockerHub username. This command will run each line in our Dockerfile to build the image of the container, then push the image to Dockerhub and create a new project called “cython_reservoir”, with the version tag 0.1.
To run the container locally, we run the command:
docker run --rm -v <working_directory>/results:/code/results <username>/cython_reservoir:0.1 mpirun -np 4 run_reservoir_sim.py
This command has several parts. First, “docker run” means to create and start a container from the image (this is like instantiating an object from a class). “—rm” means that the container can be removed after running (just the instantiated container, not the image itself). Next, the “-v” flag creates a directory mount from “code/results” inside the container to “<working_directory>/results” on your machine, where <working_directory> should be replaced with the full path of your working directory. This allows us to access the simulation results even after the container is removed. Lastly, “mpirun -np 4 run_reservoir_sim.py” tells the container to run the script across four processors – this will run four independent simulations and store each result as a csv file in the results directory.
Running on TACC clusters with Singularity
Now that we have verified that this container works on a local laptop, let’s move to Stampede2, a supercomputer owned by TACC (with allocations managed through NSF XSEDE). Unfortunately, Docker isn’t allowed on TACC’s clusters due to permission issues. However, another containerization software called Singularity is allowed, and has the ability to run containers created through Docker. After logging into the cluster through the terminal, we need to load the Singularity module:
module load tacc-singularity
We also need to create a directory called “results”. TACC doesn’t allow Singularity to be run directly from the login node, so we need to request an interactive session:
idev -m 30
When the interactive session on a compute node has been granted, we can “pull” our container from DockerHub:
Singularity creates and downloads a single file, cython_reservoir_0.1.sif, as a Singularity representation of the Docker container. Once this has downloaded, you can exit your interactive session. We will run the job itself non-interactively by submitting it to the SLURM scheduler, just like you would with a non-containerized job:
sbatch submit_reservoir_stampede2.sh
This submission script, in addition to typical SLURM directives, contains the line
mpirun singularity run cython_reservoir_0.1.sif run_reservoir_sim.py
And that’s it! This command will run the simulation across all processors assigned to the job, and store the results in the results folder. These same commands and container can also be used on the Frontera cluster (also owned by TACC), using the “submit_reservoir_frontera.sh” script. As you can see, this makes it straightforward to develop and run code across your local laptop and two different TACC clusters, without having to worry about software dependency issues.
Customizing containers to run on non-TACC clusters
Unfortunately, it’s not quite so simple to port containers to other HPC systems if your application involves MPI parallel processing. This is because it is necessary for the MPI configuration inside the container to be interoperable with the MPI configuration in the cluster’s environment. TACC’s customized base images take care of this complexity for us, but may not port to alternative systems, such as CAC’s TheCube and Hopper. In this case, we need to provide explicit instructions in the Dockerfile for how to set up MPI:
### start with ubuntu base image
FROM ubuntu:18.04
### install basics, python3, and modules needed for application
RUN apt-get update && apt-get upgrade -y && apt-get install -y build-essential zlib1g-dev libjpeg-dev python3-pip openssh-server
RUN pip3 install Pillow numpy pandas matplotlib cython
### install openMPI version 4.0.5, consistent with Hopper & TheCube
RUN wget 'https://www.open-mpi.org/software/ompi/v4.0/downloads/openmpi-4.0.5.tar.gz' -O openmpi-4.0.5.tar.gz
RUN tar -xzf openmpi-4.0.5.tar.gz openmpi-4.0.5; cd openmpi-4.0.5; ./configure --prefix=/usr/local; make all install
RUN ldconfig
### install mpi4py now that openmpi is installed
RUN pip3 install mpi4py
### add all code from current directory into “code” directory within container, and set as working directory
ADD . /code WORKDIR /code ENV PATH "/code:$PATH"
### compile cython for this particular application
RUN python3 setup.py build_ext --inplace
### set python file as executable so it can be run by docker/singularity
RUN chmod +rx /code/run_reservoir_sim.py
### change username from root
RUN useradd -u 8877 <username>
For this container, we start with a base Ubuntu image rather than TACC’s specialized image. We then install several important libraries and Python modules. The third block of commands downloads and installs OpenMPI version 4.0.5, which is the version installed on Hopper. TheCube has an older version, 3.1.4, but I found version 4.0.5 to be backwards compatible and to work on both CAC clusters. The rest of the commands are equivalent to the first Dockerfile. We can now build this second container and push it to Dockerhub as a separate version, 0.2.
We can run this container on our local machine using the same command from above, but substituting 0.2 for 0.1. Similarly, we can log onto TheCube or Hopper and execute the same commands from above, again substituting 0.2 for 0.1. Note that the singularity module on CAC clusters is “singularity” rather than “tacc-singularity”. “submit_reservoir_cube.sh” and “submit_reservoir_hopper.sh” can be used to submit to the SLURM scheduler on these systems.
Thus, while MPI does compromise container portability somewhat, it is relatively straightforward to incorporate separate builds for different HPC systems using custom Dockerfiles. With the Dockerfile in place for each system, a streamlined workflow can be developed that allows for easy project development and deployment across multiple HPC systems.
Many thanks to TACC staff for teaching me all about containers at a recent virtual workshop, and to CAC staff who helped install and troubleshoot Singularity on their system.
Hybrid parallelization is often used when performing noisy simulation-optimization on high-performance computing clusters. One common technique is to divide the available CPUs into a smaller number of “tasks”, each of which uses its subset of CPUs to assess the performance of candidate solutions through parallel evaluation of multiple scenarios. In this post, I will demonstrate how to use Python’s Multiprocessing module to set up a hybrid parallelization scheme with the Borg MOEA, and explore the resulting tradeoff between efficiency and learning rate in order to better understand when one should (and should not) implement such a scheme.
This post will bring together and build upon several previous posts on this blog, including (1) this demonstration by Andrew Dircks for how to use the Borg MOEA’s Python wrapper to optimize multi-objective simulations written in Python, using the Lake Problem as example; (2) this explanation of the Borg MOEA’s hybrid parallelization strategies by David Gold; and (3) this post by Bernardo Trindade on parallelizing Monte Carlo evaluations using Python and C/C++. The present post will give a brief overview of these topics in order to show how they can be brought together, but the reader is referred to the posts above for more detail on any of these topics.
Why hybrid parallelization?
As a motivating example, consider a complex water resources simulation model that takes 10 minutes to run a 30 year simulation. We want to explore the potential for new infrastructure investments to improve multiple objectives such as water supply reliability, cost, etc. We will try to discover high-performing solutions using an MOEA. To account for uncertainty, each candidate investment will be evaluated in 48 different stochastic scenarios.
Given the expense of our simulation model, this will be a major computational challenge. If we evaluate 20,000 candidate solutions (which is quite low compared to other studies), the total run time will be 20000 * 48 * 10 minutes = 6,667 days. Clearly, we cannot afford to run this serially on a laptop. If we have access to a computer cluster, we can parallelize the MOEA search and speed things up drastically. For example, with an XSEDE allocation on TACC Stampede2, I can run jobs with up to 128 Skylake nodes, each of which has 48 CPU, for a total of 6144 CPU per job. With this setup, we could finish the same experiment in ~1.1 days.
With the simplest possible parallelization strategy, we have a single CPU as the “Master” for the Borg MOEA, and 6143 CPU running individual function evaluations (FE). Each FE is allocated a single CPU, which runs 48 stochastic simulations in serial (i.e., there is no hybrid parallelization taking place). At the other extreme, I could set the MOEA to run with an entire node per “task”. This would allocate a full node to the Borg Master process, plus 127 nodes running individual FEs. Each FE would distribute its 48 stochastic simulations across its 48 CPUs in parallel. Thus, we are running fewer concurrent FEs (127 vs. 6143), but each finishes much faster (10 minutes vs. 480 minutes). In between these two extremes, there are intermediate parallelization schemes that allocate 2, 3, 4, 6, 8, 12, 16, or 24 CPU per task (Table 1).
So how to choose a parallelization strategy? The first thing to consider is efficiency. In theory, the scheme without hybrid parallelization (1 CPU per task) is the most efficient, since it allocates only one CPU to the Borg Master process, allowing for 6143 CPU to be simultaneously performing simulations. The 48 CPU/task scheme, on the other hand, allocates an entire node to the Borg Master (even though it only needs a single CPU to perform its duties), so that only 127*48=6096 CPU are running simulations at a given time. Thus, it should be ~1% slower in theory. However, in practice, a variety of factors such as communication or I/O bottlenecks can change this behavior (see below), so it is important to run tests on your particular machine.
If the hybrid parallelization scheme is slower, why should we even consider it? The key point is the increased potential for “learning” within the MOEA loop. Remember that the 1 CPU/task scheme has 6143 FE running concurrently, while the 48 CPU/task scheme has 127 FE. This means that the former will only have approximately 20000/6143=3.3 “generations,” while the latter will have 20000/127=157.5 generations*. Put another way, each FE will take 480 minutes (30% of total 1.1 days) to complete for the 1 CPU/task scheme, compared to 10 minutes (0.6% of total) for the 48 CPU/task scheme. The latter thus provides a more continuous stream of information to the MOEA Master, and therefore many more opportunities for the MOEA to learn what types of solutions are successful through iterative trial and error.
*Note: The Borg MOEA is a steady-state algorithm rather than a generational algorithm, so I use the term “generation” loosely to refer to the number of FE that each task will complete. See Dave Gold’s blog post on the Borg MOEA’s parallelization for more details on generational vs. steady-state algorithms. Note that for this post, I am using the master-worker variant of the Borg MOEA.
Function evaluations (FE)
Total CPU
Tasks/node
CPU/task
Concurrent FE
Generations
20000
6144
48
1
6143
3.3
20000
6144
24
2
3071
6.5
20000
6144
16
3
2047
9.8
20000
6144
12
4
1535
13.0
20000
6144
8
6
1023
19.6
20000
6144
6
8
767
26.1
20000
6144
4
12
511
39.1
20000
6144
3
16
383
52.2
20000
6144
2
24
255
78.4
20000
6144
1
48
127
157.5
Table 1: Scaling of concurrent function evaluations and generations for different hybrid parallelization schemes, using 128 Skylake nodes (48 CPU each) on Stampede2.
A simple example: hybrid parallelization of the Lake Problem in Python
Above, I have outlined a purely theoretical tradeoff between efficiency and algorithmic learning. How does this tradeoff play out in practice? What type of hybrid scheme is “best”? I will now explore these questions in more detail using a smaller-scale example. The Lake Problem is a popular example for demonstrating concepts such as non-linear dynamics, deep uncertainty, and adaptive multi-objective control; a search for “Lake Problem” on the Water Programming Blog returns many results (I stopped counting at 30). In this problem, the decision-maker is tasked with designing a regulatory policy that balances the economic benefits of phosphorus with its environmental risks, given the large uncertainty in the complex dynamics that govern lake pollution concentrations over time. In a 2017 article in Environmental Modelling & Software, Julie Quinn demonstrated the merits of Evolutionary Multi-Objective Direct Policy Search (EMODPS) for developing control policies that adaptively update phosphorus releases based on evolving conditions (e.g., current lake concentration).
A Python version of the EMODPS Lake Problem was introduced along with a Python wrapper for the Borg MOEA in this blog post by Andrew Dircks. Additionally, this post by Bernardo Trindade demonstrates how to parallelize Monte Carlo ensembles in Python as well as C/C++. For the present example, I combined aspects of these two posts, in order to parallelize the Monte Carlo evaluations within each function evaluation, using the Multiprocessing module in Python. For this blog post, I will highlight a few of the important differences relative to the non-parallel version of the problem in Andrew Dircks’ blog post, but the interested reader is referred to these previous two post for more detail. All code used to produce these results can be found in this GitHub repository.
The lake_mp.py file specifies the analysis that the Borg MOEA should run for each function evaluation – we will evaluate each candidate operating policy using an ensemble of 48 simulations, each of which uses a different 100-year stochastic realization of hydrology. This ensemble will be divided among a smaller number of CPUs (say, 16) assigned to each function evaluation. Thus, we will have to set up our code a bit differently than in the serial version. First, consider the overall “problem” function which is called by the Borg Master. Within this function, we can set up shared arrays for each objective. The Array class from the multiprocessing module allows for thread-safe access to the same memory array from multiple CPUs. Note that this module requires shared memory (i.e., the CPUs must be on the same node). If you want to share memory across multiple nodes, you will need to use the mpi4py module, rather than multiprocessing.
from multiprocessing import Process, Array
# create shared memory arrays which each process can access/write.
discounted_benefit = Array('d', nSamples)
yrs_inertia_met = Array('i', nSamples)
yrs_Pcrit_met = Array('i', nSamples)
average_annual_P = Array('d', nSamples*nYears)
Now we need to figure out which simulations should be performed by each CPU, and dispatch those simulations using the Process class from the multiprocessing module.
# assign MC runs to different processes
nbase = int(nSamples / nProcesses)
remainder = nSamples - nProcesses * nbase
start = 0
shared_processes = []
for proc in range(nProcesses):
nTrials = nbase if proc >= remainder else nbase + 1
stop = start + nTrials
p = Process(target=dispatch_MC_to_procs, args=(proc, start, stop, discounted_benefit, yrs_inertia_met, yrs_Pcrit_met, average_annual_P, natFlow, b, q, critical_threshold, C, R, newW))
shared_processes.append(p)
start = stop
# start processes
for sp in shared_processes:
sp.start()
# wait for all processes to finish
for sp in shared_processes:
sp.join()
The “args” given to the Process function contains information needed by the individual CPUs, including the shared memory arrays. Once we have appended the processes for each CPU, we start them, and then wait for all processes to finish (“join”).
The “target” in the Process function tells each CPU which subset of Monte Carlo samples to evaluate, and runs the individual evaluations in a loop:
#Sub problem to dispatch MC samples from individual processes
def dispatch_MC_to_procs(proc, start, stop, discounted_benefit, yrs_inertia_met, yrs_Pcrit_met, average_annual_P, natFlow, b, q, critical_threshold, C, R, newW):
lake_state = np.zeros([nYears + 1])
Y = np.zeros([nYears])
for s in range(start, stop):
LakeProblem_singleMC(s, discounted_benefit, yrs_inertia_met, yrs_Pcrit_met, average_annual_P, lake_state, natFlow[s, :], Y, b, q, critical_threshold, C, R, newW)
Within the loop, we call LakeProblem_singleMC to run each Monte Carlo evaluation. After running the simulation, this function can store its sub-objectives in the shared arrays:
# fill in results for MC trial in shared memory arrays, based on index of this MC sample (s)
average_annual_P[(s*nYears):((s+1)*nYears)] = lake_state[1:]
discounted_benefit[s] = db
yrs_inertia_met[s] = yim
yrs_Pcrit_met[s] = yPm
Finally, back in the main “problem” function, we can evaluate the objectives by aggregating over the shared memory arrays:
# Calculate minimization objectives (defined in comments at beginning of file)
objs[0] = -1 * np.mean(discounted_benefit) #average economic benefit
objs[1] = np.max(np.mean(np.reshape(average_annual_P, (nSamples, nYears)), axis=0)) #minimize the max average annual P concentration
objs[2] = -1 * np.sum(yrs_inertia_met) / ((nYears - 1) * nSamples) #average percent of transitions meeting inertia thershold
objs[3] = -1 * np.sum(yrs_Pcrit_met) / (nYears * nSamples) #average reliability
constrs[0] = max(0.0, reliability_threshold - (-1 * objs[3]))
time.sleep(slp)
In the last line, I add a half-second sleep after each function evaluation in order to help this example better mimic the behavior of the much slower motivating problem at the beginning of this post. Without the sleep, the Lake Problem is much faster than larger simulation codes, and therefore could exhibit additional I/O and/or master-worker interaction bottlenecks.
Again, the interested reader is referred to the GitHub repository, as well as Andrew Dircks’ original Lake Problem blog post, for more details on how these code snippets fit into the broader program.
This experiment is run on the Cube cluster at Cornell University, using 12 nodes of 16 CPU each. I tested 5 different hybrid parallelization schemes, from 1 to 16 CPU per task (Table 2). Each formulation is run for 5 seeds to account for the random dynamics of evolutionary algorithms. For this experiment, I will show results through 800 function evaluations. Although this is much smaller than the standard practice, this short experiment allows us to mimic the behavior of the much larger experiment introduced at the beginning of this post (compare the number of Generations in Tables 1 & 2).
Function evaluations (FE)
Total CPU
Tasks/node
CPU/task
Concurrent FE
Generations
800
192
16
1
191
4.2
800
192
8
2
95
8.4
800
192
4
4
47
17.0
800
192
2
8
23
34.8
800
192
1
16
11
72.7
Table 2: Scaling of concurrent function evaluations and generations for different hybrid parallelization schemes, using 12 nodes (16 CPU each) on TheCube.
How do different parallelization schemes compare?
Figure 1 shows the performance of the 1 & 16 CPU/task schemes, in terms of hypervolume of the approximate Pareto set. The hybrid parallelization scheme (16 CPU/task) is found to outperform the serial scheme (1 CPU/task). In general across the five seeds, the parallelized scheme is found to improve more quickly and to reach a higher overall hypervolume after 800 FE. The intermediate parallelization schemes (2, 4, & 8 CPU/task) are found to perform similarly to the 16 CPU/task (see GitHub repository for figures)
Figure 1: Hypervolume vs. number of function evaluations for parallelization schemes with 1 and 16 CPU per function evaluation.
Figure 2 shows the number of completed function evaluations over time, which helps illuminate the source of this advantage. The serial formulation shows a staircase pattern: large chunks of time with no progress, followed by short bursts with many FE finishing all at once. Overall, the FE arrive in 5 major bursts (with the last smaller than the rest), at approximately 25, 50, 75, 100, and 125 seconds. As we move to 2, 4, 8, and 16 CPU/task, the number of “stairs” is seen to double each time, so that the 16 CPU/task scheme almost approximates a straight line. These patterns are consistent with the generational pattern shown in Table 2.
Figure 2: Runtime vs. number of function evaluations for parallelization schemes with 1, 2, 4, 8, and 16 CPU per function evaluation.
So why does this matter? Elitist evolutionary algorithms use the relative performance of past evaluations to influence the random generation of new candidate solutions. Because a hybrid parallelization strategy evaluates fewer solutions concurrently and has more “generations” of information, there will be more ability to learn over the course of the optimization. For example, with the serial scheme, the first generation of 191 candidate solutions is randomly generated with no prior information. The 16 CPU/task scheme, on the other hand, only produces 11 solutions in the first generation, and has had ~17 generations worth of learning by the time it reaches 191 FE.
Additionally, the Borg MOEA has a variety of other auto-adaptive features that can improve the performance of the search process over the course of a single run (see Hadka & Reed, 2013). For example, Borg has six different recombination operators that can be used to mate and mutate previously evaluated solutions in order to generate new candidates for evaluation. The MOEA adaptively updates these selection probabilities in favor of the operators which have been most successful at producing successful offspring. The hybrid parallelization allows these probabilities to be adapted earlier and more regularly due to the more continuous accumulation of information (Figure 3). This can improve the performance of the MOEA.
Figure 3: Operator probabilities vs number of function evaluations (NFE) for parallelization schemes with 1 and 16 CPU per function evaluation. The subplots represent the auto-adaptive selection probabilities for the six recombination operators used by the Borg MOEA: differential evolution (DE), parent-centric crossover (PCX), simulated binary crossover (SBX), simplex crossover (PCX), uniform mutation (UM), and unimodal normal distribution crossover (UNDX).
What happens in longer runs?
At this point, it may seem that hybrid parallelization is always beneficial, since it can increase the algorithm’s adaptability (Figure 3) and improve performance (Figure 1) for a fairly negligible computational cost (Figure 2). However, the experiments in Tables 1 & 2 are very constrained in terms of the number of generations that are run. In most contexts, we will have more function evaluations and/or fewer nodes, so that the ratio of nodes to FE is much smaller and thus the number of generations is much larger. In these situations, do the advantages of hybrid parallelization hold up?
Figure 4 shows the results of a longer experiment, which is identical to above except for its use of 10,000 FE rather than 800 FE. Over the longer experiment, the advantage of the parallelized scheme (16 CPU/FE) is found to diminish, with random seed variability becoming dominant. With more generations, the differences between the two schemes seems to fade.
Figure 4: Same as Figure 1, but for a longer experiment of 10,000 function evaluations.
Figure 5 shows the evolution of the recombination operator selection probabilities over the longer experiment. The serial scheme still lags in terms of adaptation speed, but by the end of the experiment, the two formulations have very similar behavior.
Figure 5: Same as Figure 3, but for a longer experiment of 10,000 function evaluations.
Lastly, Figure 6 shows the runtime as a function of FEs for the two formulations. The parallelized scheme takes approximately 15% longer than the serial scheme. We can compare this to the theoretical slowdown based on the Borg Master overhead: the 16 CPU/task scheme runs 11 concurrent FE, each using 16 CPU, for a total of 176 CPU, while the serial version runs 191 concurrent FE, each using 1 CPU. 191 is 9% larger than 176. The difference between 15% and 9% indicates additional bottlenecks, such as writing to the shared array. This overhead may vary based on the characteristics of the cluster and the computational experiment, so testing should be performed prior to running any large experiment.
Figure 6: Same as Figure 2, but for a longer experiment of 10,000 function evaluations.
Given all this information, what to do? Should you use hybrid parallelization for your function evaluations? The answer, unsatisfyingly, is “it depends.” The most important factor to consider is the number of generations for your optimization, calculated as the total number of FE divided by the number of concurrent FE. If this number is relatively high (say, >10-20), then you may be better off sticking with serial FEs in order to improve efficiency. If the number of generations is small, it may be worthwhile to introduce hybrid parallelization in order to stimulate adaptive learning within the MOEA. Where to draw the line between these two poles will likely to vary based on a number of factors, including the number of nodes you are using, the computational architecture of the cluster, the runtime of each function evaluation, and the difficulty of the optimization problem, so testing is recommended to decide what to do in any given scenario.
This blog post is adapted from material I learned during the 2021 San Diego Supercomputer Center (SDSC) Summer Institute. This was an introductory boot camp to high-performance computing (HPC), and one of the modules taught the application of Numba for in-line parallelization and speeding up of Python code.
What is Numba?
According to its official web page, Numba is a just-in-time (JIT) compiler that translates subsets of Python and NumPy code into fast machine code, enabling it to run at speeds approaching that of C or Fortran. This is becuase JIT compilation enables specific lines of code to be compiled or activated only when necessary. Numba also makes use of cache memory to generate and store the compiled version of all data types entered to a specific function, which eliminates the need for recompilation every time the same data type is called when a function is run.
Note: The ‘@‘ flag is used to indicate the use of a decorator
Installing Numba and Setting up the Jupyter Notebook
First, in your command prompt, enter:
pip install numba
Alternatively, you can also use:
conda install numba
Next, import Numba:
import numpy as np
import numba
from numba import jit
from numba import vectorize
Great! Now let’s move onto using the @jit decorator.
Using @jit for executing functions on the CPU
The @jit decorator works best on numerical functions that use NumPy. It has two modes: nopython mode and object mode. Setting nopython=True tell the compiler to overlook the involvement of the Python interpreter when running the entire decorated function. This setting leads to the best performance. However, in the case when:
nopython=True fails
nopython=False, or
nopython is not set at all
the compiler defaults to object mode. Then, Numba will manually identify loops that it can compile into functions to be run in machine code, and will run the remaining code in the interpreter.
Here, @jit is demonstrated on a simple matrix multiplication function:
# a function that does multiple matrix multiplication
@jit(nopython=True)
def matrix_multiplication(A, x):
b = np.empty(shape=(x.shape[0],1), dtype=np.float64)
for i in range(x.shape[0]):
b[i] = np.dot(A[i,:], x)
return b
Remember – the use of @jit means that this function has not been compiled yet! Compilation only happens when you call the function:
A = np.random.rand(10, 10)
x = np.random.rand(10, 1)
a_complicated_function(A,x)
But how much faster is Numba really? To find out, some benchmarking is in order. Jupyter Notebook has a handy function called %timeit that runs simple functions many times in a loop to get their average execution time, that can be used as follows:
%timeit matrix_multiplication(A,x)
# 11.4 µs ± 7.34 µs per loop (mean ± std. dev. of 7 runs, 1 loop each)
Numba has a special .py_func attribute that effectively allows the decorated function to run as the original uncompiled Python function. Using this to compare its runtime to that of the decorated version,
%timeit matrix_multiplication.py_func(A,x)
# 35.5 µs ± 3.5 µs per loop (mean ± std. dev. of 7 runs, 10000 loops each)
From here, you can see that the Numba version runs about 3 times faster than using only NumPy arrays. In addition to this, Numba also supports tuples, integers, floats, and Python lists. All other Python features supported by Numba can be found here.
Besides explicitly declaring @jit at the start of a function, Numba makes it simple to turn a NumPy function into a Numba function by attaching jit(nopython=True) to the original function. This essentially uses the @jit decorator as a function. The function to calculate absolute percentage relative error demonstrates how this is done:
where the NumPy version takes approximately 2.61 microseconds to run, while the Numba version takes 687 nanoseconds.
Inline parallelization with Numba
The @jit decorator can also be used to enable inline parallelization by setting its parallelization pass parallel=True. Parallelization in Numba is done via multi-threading, which essentially creates threads of code that are distributed over all the available CPU cores. An example of this can be seen in the code snippet below, describing a function that calculates the normal distribution of a set of data with a given mean and standard deviation:
SQRT_2PI = np.sqrt(2 * np.pi)
@jit(nopython=True, parallel=True)
def normals(x, means, sds):
n = means.shape[0]
result = np.exp(-0.5*((x - means)/sds)**2)
return (1 / (sds * np.sqrt(2*np.pi))) * result
To appreciate the speed-up that Numba’s multi-threading provides, compare the runtime for this with:
A decorated version of the function with a disabled parallel pass
The uncompiled, original NumPy function
The first example can be timed by:
normals_deco_nothread = jit(nopython=True)(normals.py_func)
%timeit normals_deco_nothread(0.6, means, sds)
# 3.24 s ± 757 ms per loop (mean ± std. dev. of 7 runs, 1 loop each)
The first line of the code snippet first makes an uncompiled copy of the normals function, and then applies the @jit decorator to it. This effectively creates a version of normals that uses @jit, but is not multi-threaded. This run of the function took approximately 3.3 seconds.
For the second example, simply:
%timeit normals.py_func(0.6, means, sds)
# 7.38 s ± 759 ms per loop (mean ± std. dev. of 7 runs, 1 loop each)
Now, compare both these examples to the runtime of the decorated and multi-threaded normals function:
%timeit normals(0.6, means, sds)
# 933 ms ± 155 ms per loop (mean ± std. dev. of 7 runs, 1 loop each)
The decorated, multi-threaded function is significantly faster (933 ms) than the decorated function without multi-threading (3.24 s), which in turn is faster than the uncompiled original NumPy function (7.38 s). However, the degree of speed-up may vary depending on the number of CPUs that the machine has available.
Summary
In general, the improvements achieved by using Numba on top of NumPy functions are marginal for simple, few-loop functions. Nevertheless, Numba is particularly useful for large datasets or high-dimensional arrays that require a large number of loops, and would benefit from the one-and-done compilation that it enables. For more information on using Numba, please refer to its official web page.
Massively (or embarrassingly) parallel are processes that are either completely separate or can easily be made to be. This can be cases where tasks don’t need to pass information from one to another (they don’t share memory) and can be executed independently of another on whatever resources are available, for example, large Monte Carlo runs, each representing different sets of model parameters.
There isn’t any guidance on how to do this on the blog, besides an older post on how to do it using PBS, but most of our current resources use SLURM. So I am going to show two ways: a) using SLURM job arrays; and b) using the GNU parallel module. Both methods allow for tasks to be distributed across multiple cores and across multiple nodes. In terms of how it affects your workflow, the main difference between the two is that GNU parallel allows you to automatically resume/rerun a task that has failed, whereas using SLURM job arrays you have to resubmit the failed tasks manually.
Your first step using either method is to configure the function representing each task to be able to receive as arguments a task id. For example, if I would like to run my model over 100 parameter combinations, I would have to create my model function as function_that_executes_model(sample=i, [other_arguments]), where the sample number i would correspond to one of my parameter combinations and the respective task to be submitted.
For python, this function needs to be contained within a .py script which will be executing this function when called. Your .py script could look like this, using argparse to parse the function arguments but there are alternatives:
import argparse
import ...
other_arg1= 1
other_arg2= 'model'
def function_that_executes_model(sample=i, other_arg1, other_arg2):
#do stuff pertaining to sample i
return
if __name__ == '__main__':
parser = argparse.ArgumentParser(description='This function executes the model with a sample number')
parser.add_argument('i', type=int,
help='sample number')
args = parser.parse_args()
function_that_executes_model(args.i)
To submit this script (say you saved it as function_executor.py) using SLURM job arrays:
#!/bin/bash
#SBATCH --partition=compute # change to your own cluster partition
#SBATCH --cpus-per-task # number of cores for each task
#SBATCH -t 0:45:00 # max wallclock time
#SBATCH --array=1-100 # array of tasks to execute
module load python
srun python3 function_executor.py $SLURM_ARRAY_TASK_ID
This will submit 100 1-core jobs to the cluster’s scheduler, and in your queue they will be listed as JOB_ID-TASK_ID.
Alternatively, you can use your cluster’s GNU parallel module to submit this like so:
#!/bin/bash
#SBATCH --partition=compute
#SBATCH --ntasks=100
#SBATCH --time=00:45:00
module load parallel
module load python
# This specifies the options used to run srun. The "-N1 -n1" options are
# used to allocates a single core to each task.
srun="srun --export=all --exclusive -N1 -n1"
# This specifies the options used to run GNU parallel:
#
# -j is the number of tasks run simultaneously.
parallel="parallel -j $SLURM_NTASKS"
$parallel "$srun python3 function_executor.py" ::: {1..100}
This will instead submit 1 100-core job where each core executes one task. GNU parallel also allows for several additional options that I find useful, like the use of a log to track task execution (--joblog runtask.log) and --resume which will identify the last unfinished task and resume from there the next time you submit this script.
Parallel computing allows us to speed up code by performing multiple tasks simultaneously across a distributed set of processors. On high performance computing (HPC) systems, an efficient parallel code can accomplish in minutes what might take days or even years to perform on a single processor. Not all code will scale well on HPC systems however. Most code has inherently serial components that cannot be divided among processors. If the serial component is a large segment of a code, the speedup gained from parallelization will greatly diminish. Memory and communication bottlenecks also present challenges to parallelization, and their impact on performance may not be readily apparent.
To measure the parallel performance of a code, we perform scaling experiments. Scaling experiments are useful as 1) a diagnostic tool to analyze performance of your code and 2) a evidence of code performance that can be used when requesting allocations on HPC systems (for example, NSF’s XSEDE program requires scaling information when requesting allocations). In this post I’ll outline the basics for performing scaling analysis of your code and discuss how these results are often presented in allocation applications.
Amdahl’s law and strong scaling
One way to measure the performance a parallel code is through what is known as “speedup” which measures the ratio of computational time in serial to the time in parallel:
Where t_s is the serial time and t_p is the parallel time.
The maximum speedup of any code is limited the portion of code that is inherently serial. In the 1960’s programmer Gene Amdahl formalized this limitation by presenting what is now known as Amdahl’s law:
Where p is the number of processors, and s is the fraction of work that is serial.
On it’s face, Amdahl’s law seems like a severe limitation for parallel performance. If just 10% of your code is inherently serial, then the maximum speedup you can achieve is a factor of 10 ( s= 0.10, 1/.1 = 10). This means that even if you run your code over 1,000 processors, the code will only run 10 times faster (so there is no reason to run across more than 10 processors). Luckily, in water resources applications the inherently serial fraction of many codes is very small (think ensemble model runs or MOEA function evaluations).
Experiments that measure speedup of parallel code are known as “strong scaling” experiments. To perform a strong scaling experiment, you fix the amount of work for the code to do (ie. run 10,000 MOEA function evaluations) and examine how long it takes to finish with varying processor counts. Ideally, your speedup will increase linearly with the number of processors. Agencies that grant HPC allocations (like NSF XSEDE) like to see the results of strong scaling experiments visually. Below, I’ve adapted a figure from an XSEDE training on how to assess performance and scaling:
Plots like this are easy for funding agencies to assess. Good scaling performance can be observed in the lower left corner of the plot, where the speedup increases linearly with the number of processors. When the speedup starts to decrease, but has not leveled off, the scaling is likely acceptable. The peak of the curve represents poor scaling. Note that this will actually be the fastest runtime, but does not represent an efficient use of the parallel system.
Gustafson’s law and weak scaling
Many codes will not show acceptable scaling performance when analyzed with strong scaling due to inherently serial sections of code. While this is obviously not a desirable attribute, it does not necessarily mean that parallelization is useless. An alternative measure of parallel performance is to measure the amount of additional work that can be completed when you increase the number of processors. For example, if you have a model that needs to read a large amount of input data, the code may perform poorly if you only run it for a short simulation, but much better if you run a long simulation.
In the 1980s, John Gustafson proposed a relationship that notes relates the parallel performance to the amount of work a code can accomplish. This relationship has since been termed Gustafson’s law:
Where s and p are once again the portions of the code that are serial and parallel respectively and N is the number of core.
Gustafson’s law removes the inherent limits from serial sections of code and allows for another type of scaling analysis, termed “weak scaling”. Weak scaling is often measured by “efficiency” rather than speedup. Efficiency is calculated by proportionally scaling the amount of work with the number of processors and measure the ratio of completion times:
Ideally, efficiency will be close to one (the time it take one processor to do one unit of work is the same time it takes N processors to do N units of work). For resource allocations, it is again advisable to visualize the results of weak scaling experiments by creating plots like the one shown below (again adapted from the XSEDE training).
Final thoughts
Scaling experiments will help you understand how your code will scale and give you a realistic idea of computation requirements for large experiments. Unfortunately however, it will not diagnose the source of poor scaling. To improve scaling performance, it often helps to improve the serial version of your code as much as possible. A helpful first step is to profile your code. Other useful tips are to reduce the frequency of data input/output and (if using compiled code) to check the flags on your compiler (see some other tips here).
A lot of the work we do in the Reed lab involves running computational experiments on High Performance Computing (HPC) systems. These experiments often consist of performing multi-objective optimization to aid decision making in complex systems, a task that requires hundreds of thousands of simulation runs and may not possible without the use of thousands of computing core. This post will outline some best practices for performing experiments on HPC systems.
1. Have a Plan
By nature, experiments run on HPC systems will consume a large amount of computational resources and generate large amounts of data. In order to stay organized, its important to have a plan for both how the computational resources will be used and how data will be managed.
Estimating your computational resources
Estimating the scale of your experiment is the first step to running on an HPC system. To make a reasonable estimate, you’ll need to gather the following pieces of information:
How long (in wall clock time) does a single model run take on your local machine?
How many function evaluations (for an MOEA run) or ensemble model runs will you need to perform?
How long do you have in wall clock time to run the experiment?
Using this information you can estimate the number of parallel processes that you will need to successfully run the experiment. Applications such as running the Borg MOEA are known as, “embarrassingly parallel” and scale quite well with an increase in processors, especially for problems with long function evaluation times (see Hadka and Reed, 2013 for more info). However, many other applications scale poorly, so it’s important to be aware of the parallel potential of your code. A helpful tip is to identify any inherently serial sections of the code which create bottlenecks to parallelization. Parallelizing tasks such as Monte Carlo runs and MOEA function evaluations will often result in higher efficiency than paralellizing the simulation model itself. For more resources on how to parallelize your code, see Bernardo’s post from last year.
Once you have an idea of the scale of your experiment, you’ll need to estimate the experiment’s computational expense. Each HPC resource has its own charging policy to track resource consumption. For example, XSEDE tracks charges in “service units” which are defined differently for each cluster. On the Stampede2 Cluster, a service unit is defined as one node-hour of computing time, so if you run on 100 nodes for 10 hours, you spend 1,000 service units regardless of how many core per node you utilize. On the Comet Cluster, a service unit is charged by the core-hour, so if you run 100 nodes for 10 hours and each utilizes 24 core, you’ll be charged 24,000 service units. Usually, the allocations you receive to each resource will be scaled accordingly, so even though Comet looks more expensive, you likely have a much large allocation to work with. I usually make an estimate of service units I need for an experiment and add another 20% as a factor of safety.
Data management:
Large experiments often create proportionately large amounts of data. Before you start, its important to think about where this data will be stored and how it will be transferred to and from the remote system. Many clusters have limits to how much you can store on different drives, and breaking these limits can cause performance issues for the system. System administrators often don’t take kindly to these performance issues and in extreme cases, breaking the rules may result in suspension or removal from a cluster. It helps to create an informal data management plan for yourself that specifies:
How will you transfer large amounts of data to and from the cluster (tools such as Globus are helpful here).
Where will you upload your code and how your files will be structured
Where will you store data during your experimental runs. Often clusters have “scratch drives” with large or unlimited storage allocations. These drives may be cleaned periodically so they are not suitable for long term storage.
Where will you store data during post processing. This may still be on the cluster if your post processing is computationally intensive or your local machine can’t handle the data size.
Where will you store your experimental results and model data for publication and replication.
2. Test on your local machine
To make the most of your time on a cluster, its essential that you do everything you can to ensure your code is properly functioning and efficient before you launch your experiment. The biggest favor you can do for yourself is to properly test your code on a local machine before porting to the HPC cluster. Before porting to a cluster, I always run the following 4 checks:
Unit testing: the worst case scenario after a HPC run is to find out there was a major error in your code that invalidates your results. To mitigate this risk as much as possible, it’s important to have careful quality control. One helpful tool for quality control is unit testing to examine every function or module in your code and ensure it is working as expected. For an introduction to unit testing, see Bernardo’s post on Python and C++.
Memory checking: in low level code (think C, C++) memory leaks can be silent problem that throws off your results or crash your model runs. Sometimes, memory leaks can go undetected during small runs but add up and crash your system when run in large scale experiments. To test for memory leaks, make sure to use tools such as Valgrind before uploading your code to any cluster. This post features a nice introduction to Valgrind with C++.
Timing and profiling: Profile your code to see which parts take longest and eliminate unnecessary sections. If you can, optimize your code to avoid computationally intensive pieces of code such as nested loops. There are numerous tools for profiling low level codes such as Callgrind and gprof. See this post for some tips on making your C/C++ code faster.
Small MOEA tests: Running small test runs (>1000 NFE) will give you an opportunity to ensure that the model is properly interacting with the MOEA (i.e. objectives are connected properly, decision variables are being changed etc.). Make sure you are collecting runtime information so you can evaluate algorithmic performance
3. Stay organized on the cluster
After you’ve fully tested your code, it’s time to upload and port to the cluster. After transferring your code and data, you’ll likely need to use the command line to navigate files and run your code. A familiarity with Linux command line tools, bash scripting and command line editors such as vim can make your life much easier at this point. I’ve found some basic Linux training modules online that are very useful, in particular “Learning Linux Command Line” from Linked-in learning (formerly Lynda.com) was very useful.
Once you’ve got your code properly organized and compiled, validate your timing estimate by running a small ensemble of simulation runs. Compare the timing on the cluster to your estimates from local tests and reconfigure your model runs if needed. If performing an experiment with an MOEA, run a small number of NFE on a development or debug node confirm that the algorithm is running and properly parallelizing. Then run a single seed of the MOEA and perform runtime diagnostics to ensure things are working more or less as you expect. Finally, you’re ready to run the full experiment.
I’d love to hear your thoughts and suggestions
These tips have been derived from my experiences running on HPC systems, if anyone else has tips or best practices that you find useful, I’d love to hear about them in the comments.
Modern High Performance Computing (HPC) resources are usually composed of a cluster of computing nodes that provide the user the ability to parallelize tasks and greatly reduce the time it takes to perform complex operations. A node is usually defined as a discrete unit of a computer system that runs its own instance of an operating system. Modern nodes have multiple chips, often known as Central Processing Units or CPUs, which each contain multiple cores each capable of processing a separate stream of instructions (such as a single Monte Carlo run). An example cluster configuration is shown in Figure 1.
Figure 1. An example cluster configuration
To efficiently make use of a cluster’s computational resources, it is essential to allow multiple users to use the resource at one time and to have an efficient and equatable way of allocating and scheduling computing resources on a cluster. This role is done by job scheduling software. The scheduling software is accessed via a shell script called in the command line. A scheduling script does not actually run any code, rather it provides a set of instructions for the cluster specifying what code to run and how the cluster should run it. Instructions called from a scheduling script may include but are not limited to:
What code would you like the cluster to run
How would you like to parallelize your code (ie MPI, openMP ect)
How many nodes would you like to run on
How many core per processor would you like to run (normally you would use the maximum allowable per processor)
Where would you like error and output files to be saved
Set up email notifications about the status of your job
This post will highlight two commonly used Job Scheduling Languages, PBS and SLURM and detail some simple example scripts for using them.
PBS
The Portable Batching System (PBS) was originally developed by NASA in the early 1990’s [1] to facilitate access to computing resources. The intellectual property associated with the software is now owned by Altair Engineering. PBS is a fully open source system and the source code can be found here. PBS is the job scheduler we use for the Cube Cluster here at Cornell.
An annotated PBS submission script called “PBSexample.sh” that runs a C++ code called “triangleSimulation.cpp” on 128 cores can be found below:
#PBS -l nodes=8:ppn=16 # how many nodes, how many cores per node (ppn)
#PBS -l walltime=5:00:00 # what is the maximum walltime for this job
#PBS -N SimpleScript # Give the job this name.
#PBS -M email.cornell.edu # email address for notifications
#PBS -j oe # combine error and output file
#PBS -o outputfolder/output.out # name output file
cd $PBS_O_WORKDIR # change working directory to current folder
#module load openmpi/intel # load MPI (Intel implementation)
time mpirun ./triangleSimulation -m batch -r 1000 -s 1 -c 5 -b 3
To submit this PBS script via the command line one would type:
A second common job scheduler is know as SLURM. SLURM stands for “Simple Linux Utility Resource Management” and is the scheduler used on many XSEDE resources such as Stampede2 and Comet.
An example SLURM submission script named “SLURMexample.sh” that runs “triangleSimulation.cpp” on 128 core can be found below:
#!/bin/bash
#SBATCH --nodes=8 # specify number of nodes
#SBATCH --ntasks-per-node=16 # specify number of core per node
#SBATCH --export=ALL
#SBATCH -t 5:00:00 # set max wallclock time
#SBATCH --job-name="triangle" # name your job #SBATCH --output="outputfolder/output.out"
#ibrun is the command for MPI
ibrun -v ./triangleSimulation -m batch -r 1000 -s 1 -c 5 -b 3 -p 2841
To submit this SLURM script from the command line one would type:
One of my recent research tasks has been to port GCAM (Global Change Assessment Model) on to the Cube, our groups HPC cluster, and some of the TACC resources. This turned out to be more challenging than I had anticipated, so I wanted to share my experiences in the hopes that it will save you all some time in the future. This post might be a bit pedestrian for some readers, but I hope it’s helpful to folks that are new to C++ and Linux.
Before getting started, some background on GCAM. GCAM was developed by researches at the Joint Global Change Research Institute (JGCRI) at the Pacific Northwest National Lab (PNNL). GCAM is an integrated assesment model (IAM), which means it pairs a climate model with a global economic model. GCAM is unique from many IAMs in that it has a fairly sophisticated representation of various sectors of the global economy modeled on a regional level…meaning the model is a beast compared to other IAMs (like DICE). I’ll be writing a later post on IAMs more generally. GCAM is coded in C++ and makes extensive use of xml databases for both input and output. The code is open source (available here), has a Wiki (here), and a community listserve where researches can pose questions to their peers.
There are three flavors of the model available: one for Windows users, one for Mac users, and one for Unix users. The Windows version comes compiled, with a nice user-interface for post-processing the results. The Unix version comes as uncompiled code, and requires the installation of some third party C++ libraries. For those who’d like to sniff around GCAM the Windows or Mac versions are a good starting point, but if you’re going to be doing HPC heavy lifting, you’ll need to work with the Unix code.
The GCAM Wiki and the detailed user manual provide excellent documentation about running GCAM the Model Interface tools, but are a bit limited when describing how to compile the Unix version of the code. One helpful page on the Wiki can be found here. GCAM comes with a version of the Boost C++ library in the standard Unix download (located in Main_User_Workspace/libs). GCAM also uses the Berkeley DBXML library, which can be downloaded here. You’ll want to download version 2.5.16 to be sure it runs well with GCAM.
Once you’ve downloaded DBXML, you’ll need to build the library. As it turns out, this is fairly easy. Once you’ve ported the DBXML library onto your Unix cluster, simply navigate into the main directory and run the script buildall.sh. The buildall.sh script accepts flags that allow you to customize your build. We’re going to use the -b flag to build the 64-bit version of DBXML:
sh buildall.sh -b 64
once you’ve build the DBXML library, you are nearly ready to build GCAM, but must first export the paths to the Boost library and to the DBXML include and lib directories. It seems that the full paths must be declared. In other words, they should read something like:
One tricky point here is that the full path must be typed out instead of using ‘~’ as a short-cut (I’m not sure why). Once this is done, you are ready to compile gcam. Navigate into the ‘Main_User_Workspace’ directory and type the command:
make gcam
Compiling GCAM takes several minutes, but at the end there should be a gcam.exe executable file located in the ‘/exe/’ folder of the ‘Main_User_Workspace’. It should be noted that there is a multi-thread version of gcam (more info here), which I’ll be compiling in the next few days. If there are any tricks to that process, I’ll post again with some tips.