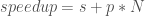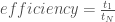In Parallel Programming Part 1, I introduced a number of basic OpenMP directives and constructs, as well as provided a quick guide to high-performance computing (HPC) terminology. In Part 2 (aka this post), we will apply some of the constructs previously learned to execute a few OpenMP examples. We will also view an example of incorrect use of OpenMP constructs and its implications.
TheCube Cluster Setup
Before we begin, there are a couple of steps to ensure that we will be able to run C++ code on the Cube. First, check the default version of the C++ compiler. This compiler essentially transforms your code into an executable file that can be easily understood and implemented by the Cube. You can do this by entering
g++ --version
into your command line. You should see that the Cube has G++ version 8.3.0 installed, as shown below.

Now, create two files:
openmp-demo-print.cppthis is the file where we will perform a print demo to show how tasks are distributed between threadsopenmp-demo-access.cppthis file will demonstrate the importance of synchronization and the implications if proper synchronization is not implemented
Example 1: Hello from Threads
Open the openmp-demo-print.cpp file, and copy and paste the following code into it:
#include <iostream>
#include <omp.h>
#include <stdio.h>
int main() {
int num_threads = omp_get_max_threads(); // get max number of threads available on the Cube
omp_set_num_threads(num_threads);
printf("The CUBE has %d threads.\n", num_threads);
#pragma omp parallel
{
int thread_id = omp_get_thread_num();
printf("Hello from thread num %d\n", thread_id);
}
printf("Thread numbers written to file successfully. \n");
return 0;
}
Here is what the code is doing:
- Lines 1 to 3 are the libraries that need to be included in this script to enable the use of OpenMP constructs and printing to the command line.
- In main() on Lines 7 and 8, we first get the maximum number of threads that the Cube has available on one node and assign it to the
num_threadsvariable. We then set the number of threads we want to use. - Next, we declare a parallel section using
#pragma omp parallelbeginning on Line 13. In this section, we are assigning theprintf()function to each of the num_threads threads that we have set on Line 8. Here, each thread is now executing theprintf()function independently of each other. Note that the threads are under no obligation to you to execute their tasks in ascending order. - Finally, we end the call to main() by returning 0
Be sure to first save this file. Once you have done this, enter the following into your command line:
g++ openmp-demo-print.cpp -o openmp-demo-print -fopenmp
This tells the system to compile openmp-demo-print.cpp and create an executable object using the same file name. Next, in the command line, enter:
./openmp-demo-print
You should see the following (or some version of it) appear in your command line:

Notice that the threads are all being printed out of order. This demonstrates that each thread takes a different amount of time to complete its task. If you re-run the following script, you will find that the order of threads that are printed out would have changed, showing that there is a degree of uncertainty associated with the amount of time they each require. This has implications for the order in which threads read, write, and access data, which will be demonstrated in the next example.
Example 2: Who’s (seg)Fault Is It?
In this example, we will first execute a script that stores each thread number ID into an array. Each thread then accesses its ID and prints it out into a text file called access-demo.txt. See the implementation below:
#include <iostream>
#include <omp.h>
#include <vector>
using namespace std;
void parallel_access_demo() {
vector<int> data;
int num_threads = omp_get_max_threads();
// Parallel region for appending elements to the vector
#pragma omp parallel for
for (int i = 0; i < num_threads; ++i) {
// Append the thread ID to the vector
data.push_back(omp_get_thread_num()); // Potential race condition here
}
}
int main() {
// Execute parallel access demo
parallel_access_demo();
printf("Code ran successfully. \n");
return 0;
}
As per the usual, here’s what’s going down:
- After including the necessary libraries, we use
include namespace stdon Line 5 so that we can more easily print text without having to used std:: every time we attempt to write a statement to the output file - Starting on Line 7, we write a simple parallel_access_demo() function that does the following:
- Initialize a vector of integers very creatively called
data - Get the maximum number of threads available per node on the Cube
- Declare a parallel for region where we iterate through all threads and
push_backthe thread ID number into thedatavector
- Initialize a vector of integers very creatively called
- Finally, we execute this function in
main()beginning Line 19.
If you execute this using a similar process shown in Example 1, you should see the following output in your command line:

Congratulations – you just encountered your first segmentation fault! Not to worry – this is by design. Take a few minutes to review the code above to identify why this might be the case.
Notice Line 15. Where, each thread is attempting to insert a new integer into the data vector. Since push_back() is being called in a parallel region, this means that multiple threads are attempting to modify data simultaneously. This will result in a race condition, where the size of data is changing unpredictably. There is a pretty simple fix to this, where we add a #pragma omp critical before Line 15.
As mentioned in Part 1, #pragma omp critical ensures that each thread executes its task one at a time, and not all at once. It essentially serializes a portion of the parallel for loop and prevents race conditions by ensuring that the size of data changes in a predictable and controlled manner. An alternative would be to declare #pragma omp single in place of #pragma omp parallel for on Line 12. This essentially delegates the following for-loop to a single thread, forcing the loop to be completed in serial. Both of these options will result in the blissful outcome of

Summary
In this post, I introduced two examples to demonstrate the use of OpenMP constructs. In Example 1, I demonstrated a parallel printing task to show that threads do not all complete their tasks at the same time. Example 2 showed how the lack of synchronization can lead to segmentation faults due to race conditions, and two methods to remedy this. In Part 3, I will introduce GPU programming using the Python PyTorch library and Google Colaboratory cloud infrastructure. Until then, happy coding!




 is the time it takes the serial version of the MOEA to achieve a given hypervolume threshold, and
is the time it takes the serial version of the MOEA to achieve a given hypervolume threshold, and 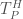 is the time it takes the parallel implementation to reach the same threshold. Figure 1 below, adapted from Hadka and Reed, (2014), shows the hypervolume speedup across different parallel implementations of the Borg MOEA for the five objective NSGA II test problem run on 16,384 processors (in this work the parallel epsilon-NSGA II algorithm is used as a baseline rather than a serial implementation). Results from Figure 1 reveal that the Multi-Master implementations of Borg are able to reach each hypervolume threshold faster than the baseline algorithm and the master-worker implementation. For high hypervolume thresholds, the 16 and 32 Master implementations achieve the hypervolume thresholds 10 times faster than the baseline.
is the time it takes the parallel implementation to reach the same threshold. Figure 1 below, adapted from Hadka and Reed, (2014), shows the hypervolume speedup across different parallel implementations of the Borg MOEA for the five objective NSGA II test problem run on 16,384 processors (in this work the parallel epsilon-NSGA II algorithm is used as a baseline rather than a serial implementation). Results from Figure 1 reveal that the Multi-Master implementations of Borg are able to reach each hypervolume threshold faster than the baseline algorithm and the master-worker implementation. For high hypervolume thresholds, the 16 and 32 Master implementations achieve the hypervolume thresholds 10 times faster than the baseline.